|
At this stage in the refinement we have a good fit for the experimental parameters, but need to improve the crystallographic model. To reduce the number of parameters needed in the initial stages of this part of the process, we will define a constraint that requires a single overall Uiso value for all atoms. |
|
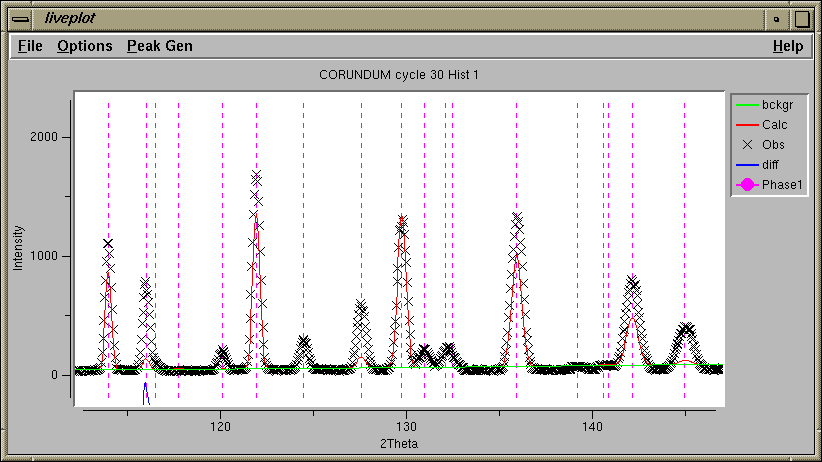
Note that at this stage in the refinement, the peak shape agrees well, but there are significant differences between the observed and computed intensities. This implies that the crystallographic model is in some way inadequate.
|
|
To group atoms so that they share a single atomic displacement parameter (displacement parameter is the preferred name for "temperature factor"), the parameters need to be constrained together. The Constraints panel allows constraints to be defined to link parameters together. At present EXPGUI allows constraints to be created for atomic parameters and for profile terms. GSAS implements many other types of constraints, but they must be accessed via the GSAS EXPEDT program. Press the "New Constraint" button at the bottom of the window to create a new constraint on atomic parameters.
While it is not needed in this case, in many projects it is best to refine an overall displacement parameter in the initial refinements for this parameter. In many refinements, there simply are not sufficient data to allow every atom to have an independently refined displacement parameter, in these circumstances, it is useful to group atoms together so that chemically similar atoms are constrained to have the same displacement parameter values. It should be noted that these constraints do not actually require that the parameters have the same value. In fact, the constraints require that the shifts that applied in future refinement cycles have their ratios defined by the constraint values. Thus, for parameters to be contrained to be equal, the parameters must start at the same value as well as have the constraint ratio set to 1. |
|
|
|
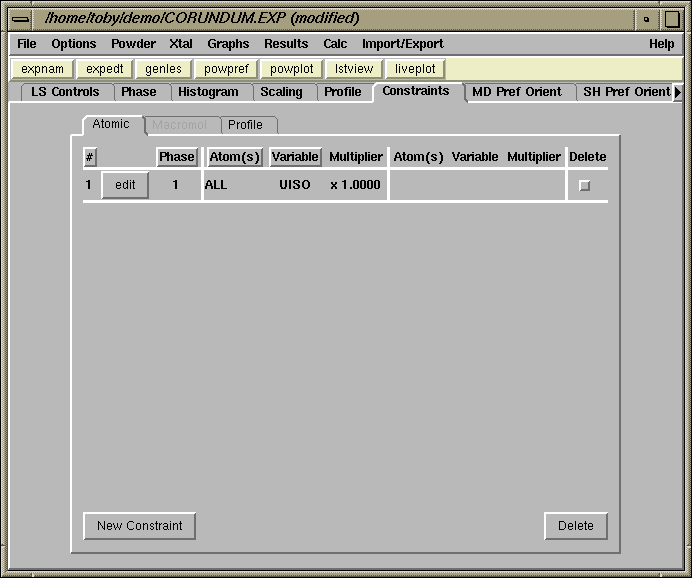
The Constraints panel now shows the constraint created in the previous step, as seen below.
|
|
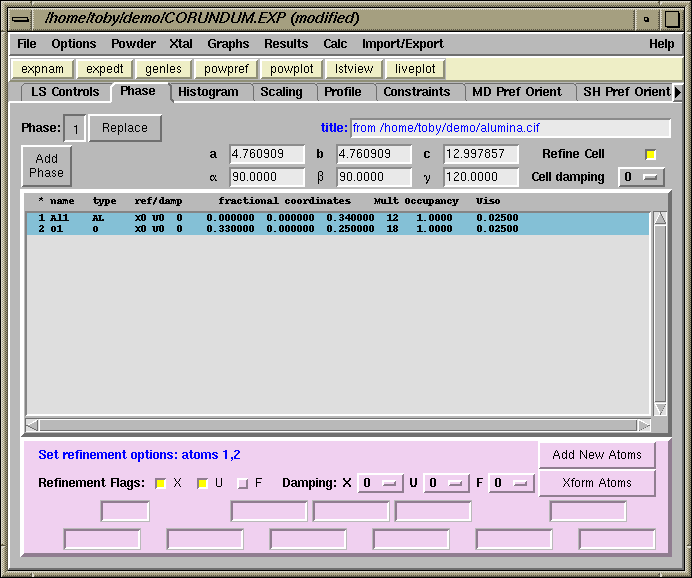
Now we are ready to set the refinement flags for the atoms on the Phase panel. Select both atoms in the list using the mouse. Select the X flag (near the bottom of the panel), this means refine x, y and z, as allowed by symmetry. Since the Al atom is on a special position, (0,0,z), only the z coordinate will be changed. Likewise the O atom is at a (x,0,1/4) position and only the x coordinate will be changed. Also select the U flag to refine the displacement (temperature) parameter for the two atoms. The previously defined constraint will be applied automatically.
|
|
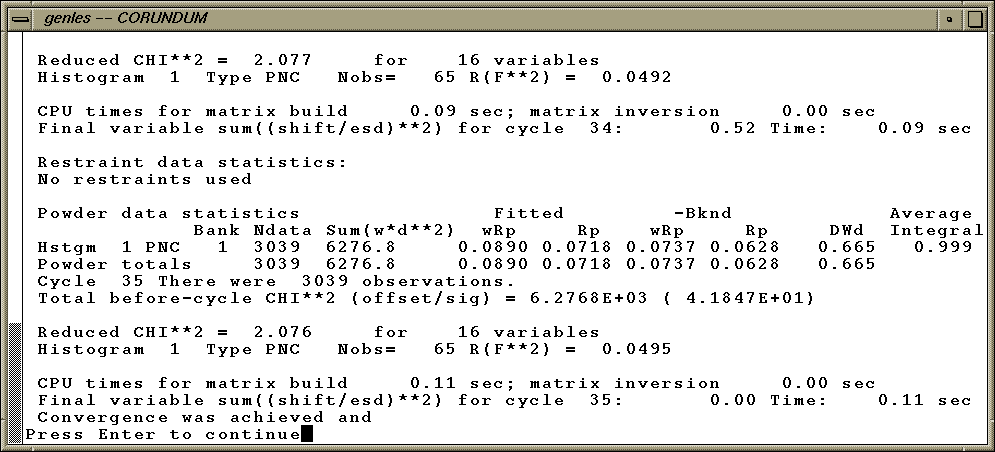
Refinement of these three atomic parameters has a enormous impact on the quality of the fit, as seen by the improvement in the agreement seen in the GENLES run below.
|
|
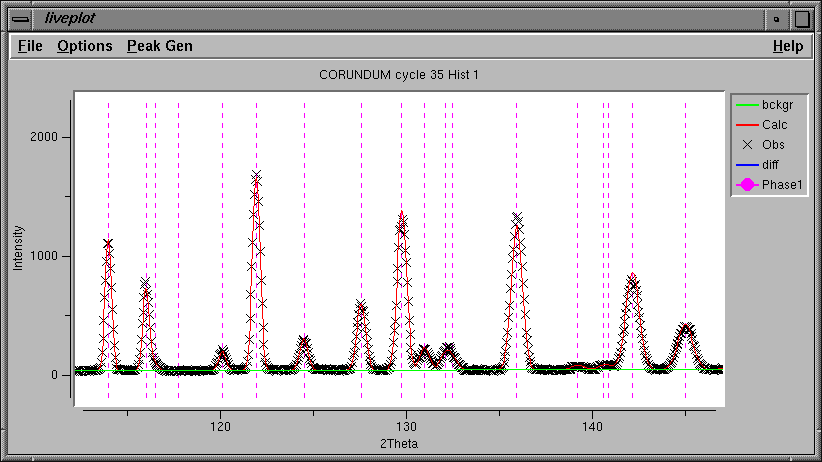
The LIVEPLOT output shows a tremendous improvement in the agreement as well.
|
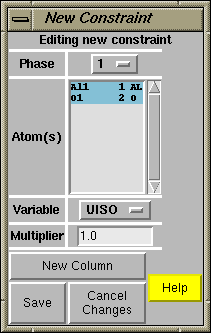 After pressing the "New Constraint" button, the window to the right is created. From top to bottom, the phase is selected as phase 1 (there is no other choice in this example), both atoms are selected (to select a range of atoms use a mouse "drag" or hold the control key while clicking the [left] mouse button; selecting all atoms can also be done by press the right mouse button. Finally select UISO for the parameter, leave the multiplier as 1 and press "Save"
After pressing the "New Constraint" button, the window to the right is created. From top to bottom, the phase is selected as phase 1 (there is no other choice in this example), both atoms are selected (to select a range of atoms use a mouse "drag" or hold the control key while clicking the [left] mouse button; selecting all atoms can also be done by press the right mouse button. Finally select UISO for the parameter, leave the multiplier as 1 and press "Save"