
Humans can learn a lot from plants. With energy from the sun, protein catalysts in plants efficiently split water to generate oxygen, storing the energy as carbohydrates. Scientists would like to perform a similar trick, using solar energy to split water and produce hydrogen fuel. Hydrogen fuel burns clean, producing only water as a byproduct, but splitting water is not an efficient task for humans. Researchers have taken baby steps toward artificial photosynthesis, building solar-powered water-splitting catalysts in the laboratory, but so far these catalysts remain far less efficient than their vegetal counterparts. One reason it's difficult to improve catalytic efficiency is that scientists don't fully understand the catalysts’ water-splitting mechanism. To examine the reaction pathway of a cobalt-based catalyst in unprecedented detail, researchers collected x-ray absorption spectra at the U.S. Department of Energy’s Advanced Photon Source (APS), an Office of Science user facility at Argonne National Laboratory. The data revealed electronic and structural features of the catalyst, offering clues for how the researchers can tweak the catalyst to run the reaction more efficiently. Such advances could bring down the cost of producing hydrogen fuel, making a hydrogen economy—a world run on water—a possibility for the future.
Photosynthetic water splitting is the breakdown of two water molecules into two H2 molecules plus an O2. The reaction takes place in two steps: 1) water oxidation and 2) proton reduction. Chemically speaking, both steps are difficult, but for this study, the researchers focused only on speeding up the second step. The tough part of proton reduction involves forming a bond between two hydrogens to form H2, a process called hydrogen evolution, so that’s where a catalyst can help. There are many hydrogen-evolving catalysts, but none rival biological enzymes, and most have additional drawbacks as well. Most existing catalysts aren’t soluble in water, which is a problem since the end goal is to use the catalyst to turn water into fuel. Another factor is cost; many catalysts rely on the electron-moving properties of expensive metals, such as platinum or rhodium. In this study, the researchers examined the reaction pathways of a water-soluble hydrogen-evolving catalyst with a lower-cost cobalt center.
The catalyst’s formula is [LCoIIICl2], with the cobalt stabilized by four nitrogen atoms embedded in a macrocycle ligand of ring moieties, represented as L in the formula. To figure out the exact steps the catalyst takes to form hydrogen-hydrogen bonds, a team of researchers from the Institute of Chemical Research of Catalonia (Spain), Southern Federal University (Russia), Universitat Autònoma de Barcelona (Spain), and Argonne used the X-ray Science Division 20-BM-B and 11-ID-D x-ray beamlines at the APS to perform static and time-resolved x-ray absorption spectroscopy (tr-XAS). As team leader Dooshaye Moonshiram explained, “The main advantages of time-resolved x-ray absorption spectroscopy over other methods is that it tracks the formation and decay of reaction intermediates, even those that disappear within picoseconds, as well as the kinetics of each reaction step. We hoped to identify the reaction’s rate-limiting step, as this is a key efficiency bottleneck and a target for optimization. “
At the beamline, the researchers mixed the cobalt catalyst with a ruthenium-based photosensitizer. This photosensitizer is energized by light and can pass that energy to the catalyst to jumpstart the reaction. They also added ascorbic acid to the mixture. The ascorbic acid acts as an electron donor, which provides a source of electrons for downstream reactions. The sample is flown into a free-flowing liquid jet sheet in the path of the x-ray beam and excited with an optical laser. The light sets the reaction in motion, with the photosensitizer funneling electrons from the donor to the catalyst. As the reaction progressed, the x-ray data showed that the structural environment around the cobalt metal center changes over the course of the reaction and also allowed the observation of the cobalt cycling through different oxidation states.
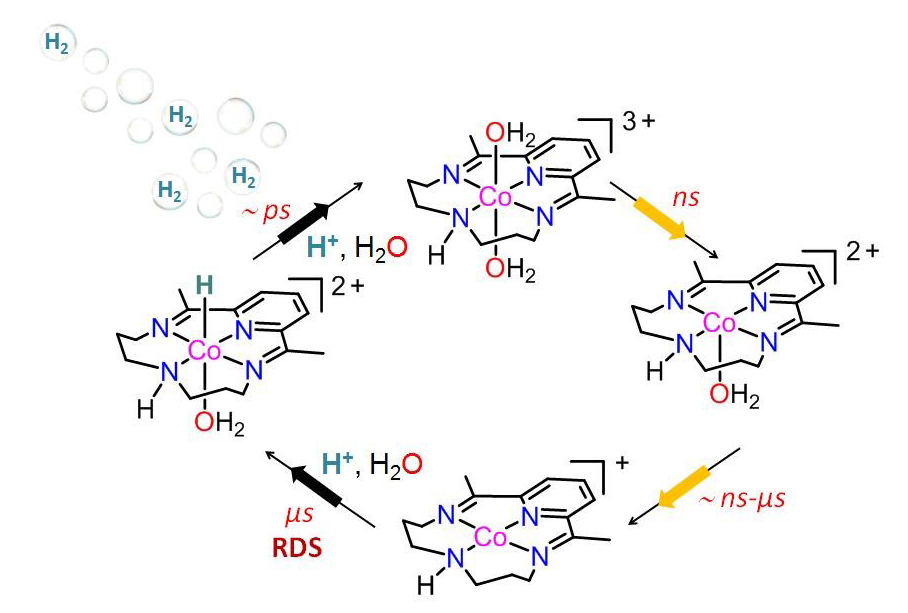
To better understand what was going on, the researchers compared the experimental data with numerical simulations of the reaction, which predicted the spectra from hypothetical intermediates. Together, the data and simulations allowed the researchers to map out the full mechanistic pathway of the reaction and determine the rate-limiting step (Fig.1). The formation of the Co(I) occurred in a matter of nanoseconds, while the formation of the Co(III) hydride intermediate took microseconds to form. The researchers concluded that the formation of the latter intermediate—the protonation of Co(I)--was therefore the rate-limiting step.
This discovery immediately suggests ways in which the researchers could adjust the structure of the macrocyclic ligand to speed up the reaction and improve the catalytic efficiency. The process is a bit of trial and error—make a tweak and see if the efficiency improves—but the researchers no longer have to make their tweaks in the dark. — Erika Gebel Berg
See: Dooshaye Moonshiram1*, Carolina Gimbert-Suriñach**2, Alexander Guda3, Antonio Picon1, C. Stefan Lehmann1, Xiaoyi Zhang1, Gilles Doumy1, Anne Marie March1, Jordi Benet-Buchholz2, Alexander Soldatov3, Antoni Llobet2,4***, and Stephen H. Southworth1, “Tracking the Structural and Electronic Configurations of a Cobalt Proton Reduction Catalyst in Water,” J. Am. Chem. Soc. 138, 10586 (2016). DOI: 10.1021/jacs.6b05680
Author affiliations: 1Argonne National Laboratory, 2Barcelona Institute of Science and Technology, 3Southern Federal University, 4Universitat Autònoma de Barcelona
Correspondence: *dmoonshi@gmail.com, **cgimbert@iciq.es, ***allobet@iciq.es
We acknowledge support from the U.S. Department of Energy (DOE) Office of Science-Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences divisions under contract no. DEAC02- 06CH11357. We also acknowledge financial support from MINECO and the “Fondo Europeo de Desarrollo Regional” (FEDER) through grants CTQ-2013−49075-R, SEV-2013-0319, and CTQ-2014-52974- REDC. Financial support from the EU COST actions CM1202 and CM1205 are also acknowledged. C.G.S. is grateful to AGAUR and Generalitat de Catalunya for a “Beatriu de Pinós” postdoctoral grant. A.G. and A.S. would like to thank the Russian Ministry of Education and Science for the support (agreement no. 14.587.21.0002, unique identifier RFMEFI58714X0002). This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit the Office of Science website.