
Researchers using an x-ray beamline at the U.S. Department of Energy’s Advanced Photon Source (APS) at Argonne National Laboratory have gained new information on how the motions of entangled polymer chains in a thin liquid film freeze as the film approaches the temperature at which the liquid goes into a glassy state.
The researchers, from the University of California, San Diego; Argonne; Sogang University; and Northern Illinois University, used the synchrotron x-ray research technique known as surface sensitive x-ray photon correlation spectroscopy (XPCS) at X-ray Operations and Research beamline 8-ID-I to investigate the dynamical relaxations of surface fluctuations of supported molten atactic polystyrene (PS) films.
Liquid surfaces are often described as being “smooth as glass.” But in fact, liquid surfaces are never smooth. They are disturbed by wind, and they are always decorated with waves caused by thermal fluctuations. At short wavelengths, these waves are governed by the surface tension and viscosity of the liquid and are called capillary waves. For highly viscous, or syrupy, liquids—in the case of this subject, polymer melts—these waves are overdamped, their amplitude reduced. Hydrodynamic theory can predict how the relaxation time depends on the wavelength of the capillary wave. This relation can in turn be used to determine the viscosity of thin liquid films.
In this experiment, the researchers measured the over-damped relaxation time of the capillary waves as a function of wavelength, molecular weight (MW), thickness, and temperature. The results showed that at high temperatures, hydrodynamic theory was very well obeyed, with liquid viscosities in good agreement with those obtained from bulk zero shear viscosities through rheological measurements.
As the films cooled down toward the glass transition temperature (Tg, the temperature at which an amorphous solid—such as glass or a polymer—becomes brittle on cooling or soft on heating), the simple exponential relaxation of the surface capillary waves was replaced by so-called “stretched exponential relaxation,” which is usually a manifestation of a distribution of relaxation times, as various dynamical modes began to freeze out. In this region, a single viscosity could not be extracted using conventional hydrodynamic theory.
However, when temperatures on the order of a few degrees above the glass transition temperature were reached, pure exponential relaxation behavior was again observed, implying that most of the other relaxation processes had frozen out on the time-scale of the measurements—except one.
Remarkably, applying hydrodynamic theory to these relaxation times yielded viscosities for the larger MW polymer chains values orders of magnitude smaller than what would have been inferred from bulk measurements of the viscosity. Equally remarkable was the discovery that these viscosities turned out to be independent of molecular weight down to a critical molecular weight (MC) of ~30,000 g/mol, which is the average molecular weight of polystyrene chains in a melt between entanglement points, or the so-called “entanglement molecular weight.”
The viscosity measured for films of all molecular weights greater than Mc at these temperatures were, as might be expected, equal to the viscosity of the liquid with chains of molecular weight equal to critical molecular weight. For polystyrene melts with chains of MW<MC, the viscosities measured with XPCS were equal to those measured in bulk rheological measurements and decreased with MW as expected.
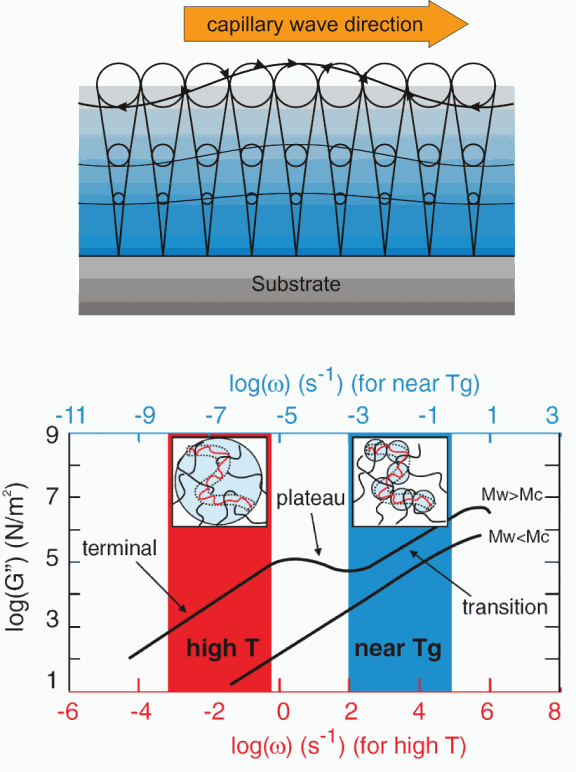
The picture that emerges from these measurements is of a gradual freezing out of the various dynamical modes of the chains, such as the famous reptation modes via which a polymer chain diffuses in a bulk melt, until at the lowest temperatures, only the modes of those portions of chains between entanglements are fluctuating (at least on the time scale of the measurements), until these too freeze out near Tg.
This interpretation is consistent with the famous “time-temperature” plots of the loss modulus G″(ω) measured for bulk polymers as a function of frequency. Capillary wave relaxation measurements carried out with XPCS probe the liquid viscosity over a given time scale (~1-1000 s) and over rather short length scales (on the order of ~nm). At temperatures well above Tg, this time scale corresponds to the low-frequency end of the so-called “terminal regime” and the viscosity is determined by the diffusion of the polymer chains along the virtual tubes formed by topological constraints, as in De Gennes’ famous reptation model. At lower temperatures, the so-called “transition regime” moves down into the window of time scales probed, and the viscosity is determined by the Rouse diffusion of the segments between entanglements only, which explains the independence of the relaxation times for MW > MC.
A universal scaling behavior was also discovered for these capillary wave relaxations. Regardless of the entanglement, thickness, or temperature, all the data are collapsed onto a simple scaling curve corresponding to a uniform density layer. This puts a strong constraint on the currently popular bilayer hypothesis, which attempts to explain the observed reduced glass transition temperature of supported polymer films by assuming a more mobile surface layer.
Conventional rheological techniques, which are usually used for bulk samples, do not have the capability to measure viscoelastic properties in film geometries. However, thermally excited surface capillary waves, which arise from the molecular arrangements throughout the entire film and therefore are encoded with hydrodynamics of the polymers, serve as a novel and unique tool to understand the viscoelastic behaviors when combined with non-invasive scattering techniques. Thus far, XPCS has proven to be the perfect technique and perhaps the only viable way to reveal these intriguing characteristics in polymer thin films.
Contact: *ZJIANG@aps.anl.gov
See: Zhang Jiang*, Mrinmay K. Mukhopadhyay, Sanghoon Song, Suresh Narayanan, L.B. Lurio, Hyunjung Kim, and Sunil K. Sinha, “Entanglement Effects in Capillary Waves on Liquid Polymer Films,” Phys. Rev. Lett. 101, 246104 (2008). DOI: 10.1103/PhysRevLett.101.246104
This work was supported by National Science Foundation Grant No. DMR-0209542, Korea Science and Engineering Foundation (KOSEF) Grants No. R01-2007-000-11808-0 and No. R15-2008-006-00000-0, and a Sogang University Research grant (2007). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America 's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.